Мукополисахаридоз 1 типа является редким, унаследованным лизосомальным расстройством хранения, которое вызывается дефицитом лизосомальных ферментов альфа-L-идуронидазы. Заболевание наследуется по аутосомно-рецессивному типу. Дефицит альфа-L-идуронидазы приводит к неспособности лизосом расщеплять дерматансульфат (DS) и гепарансульфат (GS). Этот процесс имеет важное значение для нормального роста и гомеостаза ткани. При этом заболевании, эти соединения постепенно накапливаются в лизосомах, в результате чего, клетки, ткани и органы начинают развивать разные дисфункции. На биохимическом уровне, дефицит альфа-L-идуронидазы вызывает увеличение экскреции дерматансульфата и гепарансульфата у пациентов с мукополисахаридозом 1 типа. Развитие болезни обусловлено мутацией в гене, который кодирует белок альфа-L-идуронидазу, этот ген расположен на хромосоме 4. Многие различные мутации были обнаружены в этом локусе, все эти мутации могут вызывать разные подтипы мукополисахаридоза.
Мукополисахаридоз 1 типа. Причины
Мукополисахаридоз 1 типа вызывается дефицитом фермента лизосом, L-идуронидазы. Этот недостаток приводит к накоплению мукополисахаридов, особенно дерматансульфата, в тканях и органах. Накопление избыточного сульфата дерматана приводит к постепенному развитию многочисленных морфологических аномалий в тканях и органах. Ген L-идуронидазы был обнаружен в регионе 4p16.3. Метаболический дефект имеет аутосомно-рецессивный тип наследования.
Мукополисахаридоз 1 типа. Симптомы и проявления
Исторически сложилось так, что мукополисахаридоз 1 типа можно разделить на синдром Шейе, синдром Гурлера-Шейе и на синдром Гурлера, эти разные клинические фенотипы могут проявляться от менее серьезных до наиболее тяжелых последствий для человека.
Мукополисахаридоз 1 типа. Фото

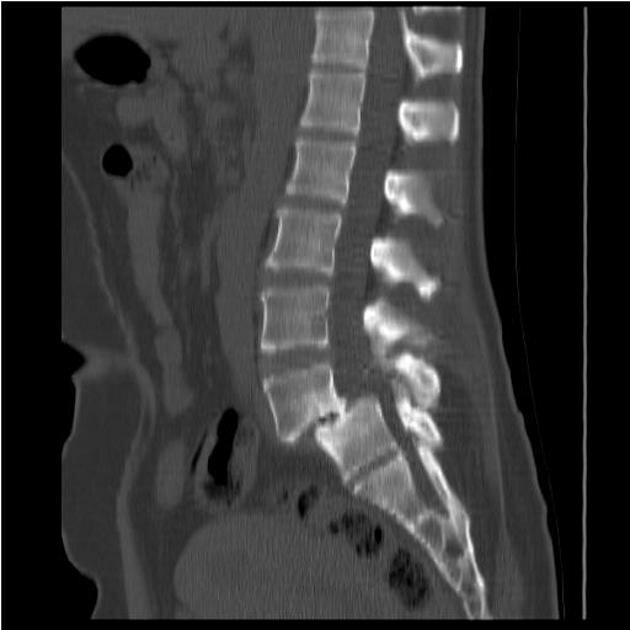
Мукополисахаридоз 1 типа. Боковая рентгенограмма грудного и поясничного отделов позвоночника.

Мукополисахаридоз 1 типа. Расширенные метафизов и диафизов.

Мукополисахаридоз 1 типа. Расширение метафизов и диафизов.

Мукополисахаридоз 1 типа.

Мукополисахаридоз 1 типа.

Мукополисахаридоз 1 типа.

Мукополисахаридоз 1 типа.
Клинические проявления мукополисахаридоза 1 типа могут проявлять хроническое мультисистемное и прогрессирующее течение. Это расстройство весьма неоднородно, оно охватывает широкий спектр тяжестей. Дети с синдромом Гурлера кажутся нормальными при рождении, но вскоре они начинают развивать характерный внешний вид. Симптомы и проявления всех типов включают развитие лицевого дисморфизма, помутнение роговицы, увеличение печени, клапанные пороки сердца, обструктивные болезни дыхательных путей, задержку развития, потерю слуха, деформации скелета и тугоподвижность суставов. Для пациентов с более тяжелой формой заболевания, наиболее типичные симптомы возникают в раннем возрасте. Эти пациенты, как правило, испытывают многочисленные изнурительные симптомы и проявления, в том числе это касается умственной отсталости. Продолжительность жизни таких людей, как правило, составляет менее 10 лет.
Однако наиболее тяжелый спектр проявлений наблюдается у пациентов с синдромом Гурлера, эти проявления также могут встречаться и при более легких синдромах, но не в таком полном наборе.
Дисморфизм лица или огрубленные черты лица
Огрубление черт лица, как правило, является первой аномалией, которая развивается у пациентов. Эти особенности часто становятся очевидными уже в возрасте 3-6 месяцев и со временем они могут постепенно становиться все более очевидными. Голова большая с выпученными лобными костями. Череп часто отличается своим большим размером от черепа здоровых детей. Спинка носа находится в депрессии, кончик носа широкий. Щеки полны. Губы увеличены, а рот обычно находится в открытом положении, особенно после возраста 3-х лет. Глаза также могут быть широко расставленными, глазницы могут быть маленькими, в результате чего глаза могут выглядеть немного выступающими. Клиническая картина менее тяжелых синдромов может быть ограничена мягким огрублением черт лица и прогнатизмом. У некоторых детей также могут быть толстые губы.
В результате дефектов хранения гликозаминогликана, у детей может развиваться прогрессивное помутнение роговицы, которое уже может начаться в первый год жизни. Помутневшая роговица имеет вид матового стекла и может привести к слепоте. Дегенерация сетчатки также является распространенным состоянием.
Очень часто, у детей диагностируется прогрессивная гепатоспленомегалия. Избытки дерматансульфата и гепарансульфата в печени и селезенке не приводят к дисфункции органов, однако, размеры органов могут быть массивными. Жидкий стул и диарея являются эпизодическими проблемами для некоторых пациентов. Паховые и пупочные грыжи также часто встречаются у пациентов. Они иногда присутствуют при рождении или развиваются в течение первых нескольких месяцев жизни и они часто являются одним из первых клинических признаков.
У больных с тяжелой болезнью, скелетные аномалии развиваются уже в раннем возрасте, примерно до возраста 6 месяцев. Расширение ребер и мягкие аномалии костей являются частыми проявлениями. На клиническом уровне, скелетные аномалии не будут вызывать дискомфорт у пациентов, пока они не достигнут возраста 10-14 месяцев. В конце концов, у детей начнут развиваться прогрессивные скелетные дисплазии с участием всех костей при всех типах синдромов. Аномалии позвонков часто приводят к деформации позвоночника. Как правило, у пациентов плохо формируется таз. Небольшие головки бедра могут привести к развитию прогрессирующих и изнурительных деформаций. Ключицы могут быть короткими, утолщенными и могут иметь странные формы (в форме весла).
Ребенок может начать испытывать боль в суставах уже в 2-летнем возрасте. Также часто встречается кистевой туннельный синдром, который, вероятно, является результатом сочетания чрезмерного хранения лизосом в соединительной ткани и деформации, вторичной по отношению к основной скелетной дисплазии.
Заболевания клапанов, в частности болезнь клапана аорты, может наблюдаться у этих больных. Частые инфекции верхних и нижних дыхательных путей также часто встречаются. Дыхательная непроходимость возникает вторично к расширению миндалин и аденоидов, хронической потере слуха и вторично к расширенному языку.
При тяжелых формах синдрома, задержка развития часто проявляется в возрасте 12-24 месяцев.
Шея короткая, иногда диагностируется сдавливание спинного мозга. Волосы на теле могут быть грубее, чем обычно, кожа может быть толще.
Мукополисахаридоз 1 типа. Диагностика
Следующие исследования показаны пациентам с подозрением на мукополисахаридоз 1 типа:
- Лимфоциты в мазках крови могут быть рассмотрены на наличие аномальных цитоплазматических структур.
- Мочевые уровни мукополисахаридов, дерматансульфата и гепарансульфата увеличиваются.
- Уровни L-идуронидазы могут быть определены в культуре фибробластов и в лейкоцитах.
- Пренатальная диагностика уровня фермента может быть также выполнена при анализе амниотических клеток и ворсинок хориона.
- В тяжелых случаях мукополисахаридоза 1 типа, рентгенография скелета (особенно позвоночника) может быть полезной для обнаружения деформаций костей позвоночника.
- Эхокардиография полезна в определении характера сердечных осложнений.
Мукополисахаридоз 1 типа. Лечение
Поскольку мукополисахаридоз 1 типа является генетическим расстройством, то назначаемые терапии будут содержать как лечебные так и паллиативные элементы. Фермент заместительная терапия лоранидазой может обеспечить клинически важные преимущества, такие как улучшение легочной функции и сокращение избыточных углеводов, хранящихся в органах.
Корректирующие операции могут быть необходимы для пациентов с мукополисахаридозом 1 типа, которые имеют контрактуры суставов, а также уродства рук и ног. Некоторым пациентам может потребоваться пересадка роговицы, если проблемы со зрением будут серьезными.
Мукополисахаридоз 1 типа. Прогноз
Симптомы более мягких типов синдромов, как правило, развиваются в конце подросткового возраста и в возрасте около 20 лет, собственно и сами симптомы и проявления будут мягче, чем симптомы, наблюдаемые при синдроме Гурлера.
Мукополисахаридоз I типа у детей
Версия: Клинические рекомендации РФ (Россия) 2013-2017
Общая информация
Краткое описание
Союз педиатров России
Клинические рекомендации: Мукополисахаридоз I типа у детей
Год утверждения (частота пересмотра): 2016 (пересмотр каждые 3 года)
Мукополисахаридозы (МПС) — группа наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов (ГАГ), приводящее к мультиорганному поражению. Обусловлены данные заболевания мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул [1-3].
Мукополисахаридоз I типа — наследственная лизосомная болезнь накопления, обусловленная дефицитом фермента альфа-L-идуронидазы и протекающая с различными клиническими проявлениями: умственной отсталостью, поражением нервной системы, сердечно-легочными нарушениями, гепато-спленомегалией, задержкой роста, множественным дизостозом, помутнением роговицы. Все вышеперечисленные признаки приводят к инвалидизации.

— Профессиональные медицинские справочники. Стандарты лечения
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS

— Профессиональные медицинские справочники
— Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID / для iOS
Классификация
E76.0 — Мукополисахаридоз 1 типа
• Мукополисахаридоз I типа (болезнь Гурлер). Шейный гиперлордоз. Правосторонний грудной сколиоз II-III степени. Килевидная деформация грудной клетки. Множественные контрактуры верхних и нижних конечностей. Вальгусная деформация нижних конечностей с вершиной на уровне коленных суставов. Плосковальгусная деформация стоп II степени. Кардиомиопатия вторичная. Миксоматоз створок митрального клапана, недостаточность митрального клапана 2 степени, недостаточность аортального клапана. НК I-IIA ст. Дисфункция синусового узла Экзофтальм, помутнение роговицы средней степени. Гиперметропия слабой степени. Птоз 1 ст. Темповая задержка речевого развития. Синдром запястного канала. Дизартрия. Пупочная грыжа.
Таблица 1 – Классификация (номенклатура) МПС

Этиология и патогенез
Эпидемиология
Клиническая картина
Cимптомы, течение
Костная система: Со стороны костно-суставной системы при МПС I выявляется множественная симптоматика. У всех пациентов формируется тугоподвижность всех групп суставов, в результате контрактур межфаланговых суставов и укорочения фаланг, образуются деформациикистей по типу «когтистой лапы». Тазобедренные суставы сформированы неправильно, головки бедренных костей маленькие, уплощенные иузурированные, характерна coxa valgum. Подвздошные кости приобретают»треугольную» деформацию. Рентгенологические изменения, видимые при синдроме Гурлер, описываются как множественный дизостоз. Для длинных трубчатых костей характерно расширение диафизов, рентгенологически неправильно проявляющиеся метафизы и эпифизы. Ключицы укорочены, утолщены. Ребра описываются как«веслообразные», их вертебральные концы сужены, а стернальные — утолщены и расширены. Фаланги кистей и стоп укорочены, имеют трапециевидную форму и расширенные диафизы. Формируются платиспондилия, кифоз, кифосколиоз. Позвонки расширены в поперечнике, высота их уменьшена. В участках, где сформирован кифоз или кифосколиоз, выявлено недоразвитие поперечных отростков позвонков или их «языкообразная» деформация.
При сдавлении спинного мозга, вызванного утолщением его оболочек или нестабильностью атлантоаксиального сустава, отмечают: нарушение походки, мышечную слабость, неуклюжесть при сохранных моторных навыках и дисфункцию мочевого пузыря.
Карпальный тоннельный синдром – частая нейропатия сдавления у пациентов в возрасте от 5 до 10 лет и взрослых. При отсутствии лечения может привести к необратимой контрактуре дистальных межфаланговых суставов, а также к нарушению или потере чувствительности первых трех пальцев и парезу мышц тенара. К сожалению, пациенты редко сообщают о болевых ощущениях, пока не происходит потеря функции.
Органы дыхания: частые респираторные заболевания в виде ринитов, отитов. Накопление ГАГ в миндалинах, надгортаннике, а также в трахее приводит к утолщению и сужению дыхательных путей и развитию обструктивного апноэ.
Также встречаются: гипертрихоз, гепатоспленомегалия, нарушение слуха, пупочная и/или паховая грыжи. На поздних стадиях у детей выявляют тугоухость, снижение зрения и умственную отсталость .
Желудочно-кишечная система: гепатоспленомегалия, пахово-мошоночные и пупочные грыжи.
Диагностика
Диагноз МПС I устанавливается на основании совокупности клинических данных, результатов лабораторного исследования и молекулярно-генетического анализа. Частота применения методов при первоначальной оценке и дальнейшем наблюдении приведена в Приложении Г2 [1-3].
Мукополисахаридоз, тип I. Причины. Симптомы. Диагностика. Лечение

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Мукополисахаридоз, тип I (Синонимы: недостаточность фермента лизосомной a-L-идуронидазы, синдромы Гурлер, Гурлер-Шейе и Шейе).
Мукополисахаридоз, тип I — аутосомно-рецессивное заболевание, возникающее в результате снижения активности лизосомной a-L-идуронидазы, которая участвует в метаболизме гликозаминогликанов. Заболевание характеризуется прогрессирующими нарушениями со стороны внутренних органов, костной системы, психоневрологическими и сердечно-лёгочными расстройствами.
- Е76 Нарушения обмена гликозаминогликанов.
- Е76.0 Мукополисахаридоз, тип I.
Мукополисахаридоз I — панэтническое заболевание с частотой встречаемости в популяции в среднем 1 на 90 000 живых новорождённых. Средняя частота синдрома Гурлер в Канаде 1 на 100 000 живых новорождённых, синдрома Гурлер-Шейе — 1 на 115 000, синдрома Шейе — 1 на 500 000.
В зависимости от выраженности клинических симптомов заболевания различают три формы мукополисахаридоза I: синдромы Гурлер, Гурлер-Шейе и Шейе.
Причины мукополисахаридоза I типа
Мукополисахаридоз I — аутосомно-рецессивное заболевание, возникающее в результате мутаций в структурном гене лизосомной альфа-L-идуронидазы.
Ген альфа-L-идуронидазы — IDUA — расположен на коротком плече хромосомы 4 в локусе 4р16.3. К настоящему времени известно более 100 различных мутаций в гене IDUA. Превалирующее число известных мутаций — точечные в различных экзонах гена IDUA. Для европеоидов характерны две частые мутации Q70X и W402X.
Самая распространённая мутация среди пациентов из российской популяции — мутация Q70X. Её частота — 57%, что сравнимо с частотой Q70X в скандинавской популяции (62%). Частота мутации W402X, которая встречается в 48% случаев мукополисахаридоза I в ряде европейских популяций, в российской популяции составляет 5,3%.
Патогенез мукополисахаридоза I типа
Фермент a-L-идуронидаза участвует в метаболизме двух гликозаминогликанов — дерматан-сульфата и гепарансульфата. Поскольку идуроновая кислота входит в состав дерматансульфата и гепарансульфата, при данном заболевании нарушен внутрилизосомный распад именно этих гликозаминогликанов, которые и накапливаются в лизосомах повсеместно: в хрящах, сухожилиях, надкостнице, эндокарде и сосудистой стенке, печени, селезёнке и нервной ткани. Отёк мягкой мозговой оболочки вызывает частичную окклюзию субарахноидальных пространств, что приводит к прогрессирующей внутренней и наружной гидроцефалии.
Поражаются клетки коры большого мозга, таламуса, ствола, передних рогов. Тугоподвижность суставов — результат деформации метафизов, утолщение суставной капсулы вторично по отношению к отложению в ней гликозаминогликанов и фиброзу. Обструкция дыхательных путей — следствие сужения трахеи, утолщения голосовых связок, избыточности отёчных тканей в верхних дыхательных путях.
Симптомы мукополисахаридоза I типа
Мукополисахаридоз, тип IH (синдром Гурлер)
У больных с синдромом Гурлер первые клинические признаки заболевания появляются на первом году жизни, с пиком манифестации от 6 до 12 мес. В ряде случаев уже с рождения наблюдают незначительное увеличение печени, пупочные или пахово-мошоночные грыжи. Обычно диагноз устанавливают в возрасте от 6 до 24 мес. Характерные изменения черт лица по типу гаргоилизма становятся очевидными к концу первого года жизни: большая голова, выступающие лобные бугры, широкая переносица, короткие носовые ходы с вывернутыми наружу ноздрями, полуоткрытый рот, большой язык, толстые губы, гиперплазия дёсен, нерегулярные зубы. Другие частые манифестные симптомы — тугоподвижность мелких и крупных суставов, кифоз поясничного отдела позвоночника (поясничный гибус), хронические отиты и частые инфекционные заболевания верхних дыхательных путей. Практически у всех пациентов с синдромом Гурлер, так же как и при других типах мукополисахаридоза, кожные покровы плотные на ощупь. Часто встречается гипертрихоз. У единичных больных в возрасте до 1 года заболевание дебютировало с развития острой сердечной недостаточности, вызванной эндокардиальным фиброэластозом. По мере прогрессирования заболевания присоединяются симптомы, свидетельствующие о вовлечении в патологический процесс внутренних органов, сердечно-лёгочной, центральной и периферической нервной систем. Ведущие неврологические симптомы — снижение интеллекта, задержка речевого развития, изменения мышечного тонуса, сухожильных рефлексов, поражение черепных нервов, комбинированная кондуктивная и нейросенсорная тугоухость. Прогрессирующая вентрикуломегалия часто приводит к развитию сообщающейся гидроцефалии. К концу первого и в начале второго года жизни появляются шумы в сердце, позднее формируются приобретённые аортальные и митральные пороки сердца. К концу второго года жизни выявляют гепатоспленомегалию и характерные скелетные нарушения по типу множественного дизостоза: короткую шею, задержку роста, тотальную платиспондилию, поясничный гибус, тугоподвижность мелких и крупных суставов, дисплазию тазобедренных суставов, вальгусную деформацию суставов, изменение со стороны кистей по типу «когтистой лапы», деформацию грудной клетки в виде бочкообразной или колоколообразной. Часто наблюдается прогрессирующее помутнение роговицы, мегалокорнеа, глаукома, застойные диски зрительных нервов и/ или их частичная атрофия.
Ранние рентгенологические признаки — деформация рёбер (по типу «вёсельных») и овоидная деформация тел позвонков, излишняя трабекуляция диафизов длинных трубчатых костей в сочетании с её недостаточностью в области метафизов и эпифизов. По мере прогрессирования заболевания формируется макроцефалия с утолщением костей свода черепа, преждевременным закрытием лямбдовидного и сагиттального швов черепа, уменьшение орбит, расширение спинки турецкого седла. Больные погибают обычно в возрасте до 10 лет от обструкции дыхательных путей, респираторных инфекций, сердечной недостаточности.
Мукополисахаридоз, тип I-H/S (синдром Гурлер-Шейе) Клинический фенотип синдрома Гурлер-Шейе занимает промежуточное положение между синдромами Гурлер и Шейе, для него характерны медленно прогрессирующие нарушения со стороны внутренних органов, костной системы, лёгкое снижение интеллекта или отсутствие такового. Заболевание обычно дебютирует в возрасте 2-4 лет. Основные клинические нарушения — поражение сердца и развитие обструктивного синдрома верхних дыхательных путей. У некоторых пациентов наблюдают тотальный спондилолистез, что может приводить к компрессии спинного мозга. Большинство пациентов доживают до третьего десятилетия жизни. Основная причина летального исхода — острая сердечно-сосудистая и лёгочная недостаточность.
Мукополисахаридоз, тип IS (синдром Шейе)
В первоначальной классификации мукополисахаридозов, до открытия первичного биохимического дефекта при синдроме Шейе, его выделяли в отдельный тип — мукополисахаридоз V. Синдром Шейе — наиболее мягкий по течению заболевания среди других форм мукополисахаридоза I, для него характерны тугоподвижность суставов, аортальные пороки сердца, помутнение роговицы и признаки множественного костного дизостоза. Первые симптомы обычно появляются в возрасте от 5 до 15 лет. Ведущие клинические симптомы — скелетные нарушения в виде тугоподвижности суставов с развитием карпального туннельного синдрома. Офтальмологические расстройства включают помутнение роговицы, глаукому и пигментную дегенерацию сетчатки. Нейросенсорная тугоухость — позднее осложнение заболевания. Обструктивный синдром верхних дыхательных путей часто приводит к возникновению апноэ во время сна, что в некоторых случаях требует установления трахеостомы. Миелопатия шейного отдела спинного мозга встречается реже, чем при синдроме Гурлер-Шейе. Часто отмечают стеноз аорты с недостаточностью кровообращения и гепатоспленомегалию. Интеллект при данном синдроме не страдает или наблюдают лёгкие когнитивные нарушения.
Диагностика мукополисахаридоза I типа
Подтверждающая биохимическая диагностика мукополисахаридоза I заключается в определении уровня экскреции гликозаминогликанов мочи и измерении активности лизосомной a-L-идуронидазы. Суммарная экскреция Ггликозаминогликанов в моче возрастает. Также наблюдают гиперэкскрецию дерматансульфата и гепарансульфата. Активность a-L-идуронидазы измеряется в лейкоцитах или культуре кожных фибробластов с использованием искусственного флюорогенного или хромогенного субстратов.
Пренатальная диагностика возможна путём измерения активности a-L-идуронидазы в биоптате ворсин хориона на 9-11-й неделе беременности и/или определения спектра ГАГ в амниотической жидкости на 20-22-й неделе беременности. Для семей с известным генотипом возможно проведение ДНК-диагностики.
При рентгенографии у пациентов с синдромом Гурлер обнаруживают типичные признаки так называемого множественного костного дизостоза. При МРТ головного мозга обнаруживают множественные кисты в перивентрикулярных областях белого вещества головного мозга, мозолистого тела, реже базальных ганглиев, признаки гидроцефалии; в редких случаях — пороки головного мозга в виде лиссэнцефалии, мальформации Денди-Уокера.
Дифференциальная диагностика проводится как внутри группы мукополисахаридозов, так и с другими лизосомными болезнями накопления: муколипи-дозами, галактосиалидозом, сиалидозом, маннозидозом, фукозидозом, GM1-ганглиозидозом.
Лечение мукополисахаридоза I типа
При синдроме Гурлер показана трансплантация костного мозга, которая может кардинально изменить течение заболевания и улучшить его прогноз, однако эта процедура имеет много осложнений и проводится на ранних стадиях заболевания, преимущественно в возрасте до 1,5 лет. В настоящее время создан препарат для ферментозаместительной терапии мукополисахаридоза I — альдуразим (Aldurazyme, Genzyme), который зарегистрирован в странах Европы, США, Японии; его применяют для лечения экстраневральных нарушений при мукополисахаридозе I. Препарат показан для коррекции мягких форм мукополисахаридоза I (синдромах Гурлер-Шейе и Шейе). Препарат вводят еженедельно, внутривенно, капельно, медленно, в дозе 100 ЕД/кг. Для лечения синдрома Гурлер с выраженными неврологическими осложнениями препарат менее эффективен, поскольку фермент не проникает через гематоэнцефалический барьер.
 [1], [2]
[1], [2]
Мукополисахаридоз
Мукополисахаридозы – это группа генетически обусловленных заболеваний, возникающих вследствие нарушения обмена кислых мукополисахаридов (гликозаминогликанов). Характерны системные поражения скелета и задержка физического развития. При некоторых формах наблюдается умственная отсталость. Возможны нарушения сердечной деятельности, патология органов зрения, образование грыж, неврологические нарушения, гипертрихоз, увеличение печени и селезенки. Диагноз выставляется на основании клинических признаков, данных рентгенографии и других исследований. Лечение симптоматическое.
МКБ-10
- Виды мукополисахаридоза
- Мукополисахаридоз типа IH
- Мукополисахаридоз типа I-S
- Мукополисахаридоз типа II
- Мукополисахаридоз типа III
- Мукополисахаридоз типа IV
- Другие типы мукополисахаридоза
- Диагностика
- Лечение мукополисахаридоза
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Мукополисахаридозы – группа генетических заболеваний, сопровождающихся накоплением кислых мукополисахаридов в органах и тканях. Причиной развития является передающаяся по наследству неполноценность лизосомных ферментов. Впервые мукополисахаридоз был описан Гурлер в 1917 году. Лечение мукополисахаридоза осуществляют травматологи-ортопеды при участии кардиологов, офтальмологов, неврологов, отоларингологов и других специалистов.
Виды мукополисахаридоза
Мукополисахаридоз типа IH
Мукополисахаридоз типа IH или синдром Гурлер встречается у 1 новорожденного из 20-25 тысяч. Симптомы мукополисахаридоза появляются в течение первого года жизни, полная клиническая картина формируется к 1-2 годам. Для данной формы мукополисахаридоза характерны грубые черты лица и деформация черепа в форме лодочного киля (скароцефалия). Из-за увеличенных аденоидов и пороков развития в области лица и носа больные дышат ртом. Отмечаются прогрессирующие деформации конечностей и других частей скелета, отставание в росте.
Позвоночник пациентов с мукополисахаридозом искривлен, из-за чего в положении сидя возникает симптом «кошачьей спины». Отмечается укорочение шеи, высокое расположение лопаток и выстояние нижних ребер. Кисти широкие, напоминающие когтистую лапу. Со временем у больных мукополисахаридозом формируются контрактуры суставов. Вначале поражаются локтевые и плечевые суставы, затем – коленные, тазобедренные и голеностопные. Из-за ограничения движений возникает характерная походка – на цыпочках с полусогнутыми ногами.
Тоны сердца приглушены, границы расширены. При аускультации определяются систолические шумы, на ЭКГ выявляется диффузное поражение миокарда. Передняя брюшная стенка пациентов с мукополисахаридозом ослаблена, живот увеличен, часто выявляются гидроцеле, пупочные и паховые грыжи. Печень и селезенка увеличены. Характерны нарушения зрения и слуха. Возможно помутнение и увеличение роговицы, пигментная дистрофия сетчатки, глаукома, атрофия зрительных нервов и застойные явления в области глазного дна. У больных мукополисахаридозом этого типа развивается прогрессирующая умственная отсталость. Могут возникать нарушения координации движений, парезы и параличи. Выявляется избыточный рост пушковых волос.
Рентгенография позвоночника пациентов с мукополисахаридозом свидетельствует о характерной деформации позвонков. Позвонки имеют кубовидную форму, их контуры закруглены, в переходном отделе выявляется скошенность передневерхних углов, углообразный кифоз, утолщение и укорочение отростков. На рентгенографии грудной клетки определяется утолщение передних и истончение задних отделов ребер, сопровождающиеся их лопатовидной или саблевидной деформацией, укорочение и деформация ключиц, уменьшение и смещение головок плечевых костей.
Рентгенография таза подтверждает сужение тазового кольца, скошенность вертлужных впадин и уменьшение головок бедренных костей. На рентгенограммах трубчатых костей выявляется истончение кортикального слоя и расширение костномозгового канала. Рентгенография костей кисти свидетельствует о недоразвитии ногтевых фаланг, укорочении и расширении пястных костей, проксимальных и средних фаланг. На рентгенограммах черепа определяется недоразвитие костей лицевого черепа, краниостеноз и макроцефалия.
Мукополисахаридоз типа I-S
Мукополисахаридоз типа I-S или болезнь Шейе (описана в 1962 г. американским офтальмологом Шейе) является более поздним, относительно благоприятно протекающим вариантом мукополисахаридоза типа IH (синдрома Гурлер). До 3-6 лет развитие детей соответствует норме. Первым признаком мукополисахаридоза становятся сгибательные контрактуры пальцев рук. В последующем ограничивается разгибания в лучезапястных, локтевых и плечевых суставах. Контрактуры нижних конечностей, как правило, слабо выражены. Полная клиническая картина мукополисахаридоза формируется к началу подросткового возраста.
Пациенты с мукополисахаридозом коренастые, невысокие, с грубыми чертами лица и хорошо развитой мускулатурой. Отмечается повышенное оволосение (гипертрихоз). Часто возникают паховые или пупочные грыжи. Кожа на пальцах натянута и утолщена. Из-за сдавления срединного нерва возможно развитие синдрома запястного канала, сопровождающееся атрофией мышц области тенара и парестезиями в области III-IV пальцев. У некоторых больных мукополисахаридозом выявляется аортальный стеноз, недостаточность клапанов аорты, пигментная дистрофия сетчатки, глаукома и помутнение роговицы. Интеллект в норме, увеличение селезенки и печени нехарактерно. На рентгенограммах определяется картина, аналогичная мукополисахаридозу типа IH, но патологические изменения выражены менее резко.
Мукополисахаридоз типа II
Мукополисахаридоз типа II или синдром Хантера чаще выявляется у мальчиков и обычно развивается на 2-3 году жизни. Как и при мукополисахаридозе типа IH наблюдается скафоцефалия, огрубление черт лица, низкий голос и затруднения дыхания из-за деформации лицевого скелета. При этом характерный для мукополисахаридоза типа IH кифоз обычно не выявляется, симптом «кошачьей спины» отрицательный. Пациенты часто страдают от респираторных инфекций (бронхитов, трахеитов, пневмоний). Постепенно развиваются нарушения координации движений, возрастает агрессивность. Настроение неустойчивое, с резкими перепадами.
Также при данной форме мукополисахаридоза наблюдается незначительное увеличение селезенки и печени, прогрессирующая тугоухость, узелки на коже спины и некоторое снижение интеллекта. В последующем может развиться помутнение роговицы. Рентгенологическая картина – как при мукополисахаридозе типа I-S. Возможны два варианта развития болезни: благоприятный (вариант В) и неблагоприятный (вариант А). При благоприятном варианте симптомы выражены нерезко, иногда возникает незначительная умственная отсталость, пациенты с мукополисахаридозом могут доживать до 30 и более лет. Для неблагоприятного варианта характерна яркая клиническая картина и серьезные нарушения интеллекта. Летальный исход наступает в подростковом возрасте.
Мукополисахаридоз типа III
Мукополисахаридоз типа III или болезнь Санфилиппо (описана в 1963 г. американским педиатром Санфилиппо) развивается у 1 новорожденного из 100-200 тысяч. В первые годы жизни развитие соответствует возрасту, иногда наблюдаются затруднения глотания и неуклюжая походка. В возрасте 3-5 лет ребенок с мукополисахаридозом становится апатичным и начинает отставать в развитии. Возникают нарушения речи, огрубление черт лица, недержание мочи и кала. Со временем умственная отсталость прогрессирует и занимает центральное положение в клинической картине этого типа мукополисахаридоза.
Наряду с нарушениями интеллекта наблюдается умеренное увеличение печени и селезенки, повышенное оволосение, контрактуры и задержка роста. Патология со стороны глаз и сердечно-сосудистой системы для этой формы мукополисахаридоза нехарактерна. Рентгенологическая картина – без изменений или как при мукополисахаридозе типа I-S, но менее выраженная. Больные мукополисахаридозом третьего типа, как правило, погибают в возрасте 10-20 лет от инфекционных осложнений.
Мукополисахаридоз типа IV
Мукополисахаридоз типа IV или болезнь Моркио (описана в 1929 г. уругвайским педиатром Моркио) наблюдается у 1 новорожденного из 40 тысяч. До 1-3 лет дети развиваются нормально. В последующем возникает значительное отставание в росте, укорочение шеи и туловища, контрактуры и вальгусная деформация конечностей, сколиоз или кифоз, разнообразные деформации грудной клетки, снижение силы мышц, утолщение кожи и огрубление черт лица. При этом типе мукополисахаридоза часто развиваются паховые и пупочные грыжи, дистрофия роговицы и тугоухость. Интеллект сохранен.
На рентгенограммах позвоночника определяется кифоз, сколиоз, расширение и уплощение тел позвонков. При проведении рентгенографии таза и конечностей выявляются множественные деформации, неровность контуров, уплощение головок бедренных костей, укорочение костей предплечья и деформации стоп. Средняя продолжительность жизни больных мукополисахаридозом – менее 20 лет. Смерть при данной форме мукополисахаридоза наступает из-за сопутствующих заболеваний, осложняющихся сердечно-легочной недостаточностью.
Другие типы мукополисахаридоза
Мукополисахаридоз типа VI или болезнь Марото-Лами (описана в 1960 г. французами Лами и Марото) развивается в возрасте 2 года и старше. Возникает огрубление черт лица, отставание в росте, укорочение шеи, контрактуры суставов и бочкообразная деформация грудной клетки. Характерны частые простуды. Возможны грыжи, увеличение печени и селезенки. Интеллект не страдает. Наблюдается два варианта течения: классический и мукополисахаридоз со слабой выраженностью клинических проявлений. На рентгенограммах больных мукополисахаридозом определяется кубовидная или клиновидная деформация позвонков, треугольная деформация таза, недоразвитие и деформация головок бедренных костей, укорочение малоберцовых костей.
Мукополисахаридоз типа VII протекает, как мупоколисахаридоз типа III, различия выявляются только при проведении биохимических исследований. Мукополисахаридоз типа VIII по симптомам напоминает мукополисахаридоз типа IV, но, в отличие от него, сопровождается задержкой умственного развития.
Диагностика
Диагноз мукополисахаридоза устанавливается на основании характерной клинической и рентгенологической картины, выявлении гликозаминогликанов в моче и изучении активности ферментов в клеточных культурах. В ходе обследования больным мукополисахаридозом назначаются консультации различных специалистов: кардиолога, гастроэнтеролога, офтальмолога, отоларинголога, невролога, психиатра и т. д., проводятся инструментальные исследования для оценки состояния различных органов и систем.
Лечение мукополисахаридоза
Патогенетическая терапия не разработана. Лечение симптоматическое, может быть как консервативным, так и оперативным. Осуществляется профилактика и лечение респираторных инфекций, проводится коррекция нарушений зрения и слуха. При необходимости выполняются грыжесечение и герниопластика, операции по устранению контрактур и коррекции деформаций скелета.
Прогноз и профилактика
Прогноз при всех типах мукополисахаридоза неблагоприятный – продолжающееся накопление продуктов обмена в тканях приводит к усугублению патологических изменений со стороны всех органов и систем. Использование любых лечебных средств (переливания крови, введение гормонов и т. д.) при мукополисахаридозе обеспечивает лишь временное улучшение. Рекомендуется пренатальная профилактика.
Мукополисахаридоз
Мукополисахаридоз – наследственное заболевание соединительной ткани, связанное с нарушением обмена гликозаминогликанов, для которого характерен специфический внешний вид лица, аномалии глаз, костей, селезенки, печени. В некоторых случаях заболевание может сопровождаться задержкой умственного развития.
Существуют различные типы мукополисахаридоза, развитие каждого из которых обусловлено определенной генетической аномалией, которая оказывает влияние на синтез специфического фермента.
Существующие методы лечения мукополисахаридоза имеют низкий уровень эффективности.
Причины мукополисахаридоза
Причиной развития мукополисахаридоза является нарушение ферментативного катализа гликозаминогликанов в лизосомах. При мукополисахаридозе нарушается процесс расщепления и сохранения мукополисахаридов, которые являются основными компонентами соединительной ткани. Избыток мукополисахаридов проникает в кровь и накапливается в тканях. Поэтому данное заболевание относят к болезням накопления.
В настоящее время выявлено 10 генетических типов мукополисахаридоза, четыре из которых возникают при нарушении активности гликозидаз, пять – сульфатаз, один развивается в случае дефицита трансферазы.
Заболевание наследуется по аутосомно-рецессивному и рецессивному, сцепленному с Х-хромосомой типам.
Типы и симптомы мукополисахаридоза
Выделяют следующие типы мукополисахаридоза:
- I тип или синдром Гурлера;
- II тип или синдром Гунтера;
- III тип или синдром Санфилиппо;
- IV тип или синдром Моркио;
- V тип или синдром Шейе;
- VI тип или синдром Марото-Лами;
- VII тип или синдром Слая;
- VIII тип или синдром Ди Ферранте.
Симптомы мукополисахаридоза обнаруживаются у ребенка на первом году жизни и уже к 12-24 месяцам они становятся довольно выраженными.
У таких детей наблюдаются:
- Череп в форме лодочного киля;
- Шумное дыхание ртом;
- Грубые черты лица;
- Отставание в росте;
- Деформации скелета;
- Постепенное развитие сгибательных контрактур;
- Увеличение размеров живота;
- Пупочные и паховые грыжи;
- Гидроцеле;
- Изменения со стороны глаз: помутнение и увеличение размеров роговицы, врожденная глаукома, атрофия сосков зрительных нервов, застойные явления на глазном дне, пигментная дистрофия сетчатки;
- Снижение слуха;
- Изменения со стороны сердца;
- Умственная отсталость;
- Повышенный тонус мышц, нарушение координации движений, параличи.
При I типе заболевания симптомы мукополисахаридоза при рождении отсутствуют. Первые симптомы мукополисахаридоза в виде ограниченного разгибания пальцев на руках наблюдаются только в 3-6 лет. Со временем уменьшается объем движений в других суставах рук. К периоду полового созревания все симптомы проявляются особенно ярко.
II тип мукополисахаридоза чаще встречается у мальчиков. Для больных характерны: скафоцефалия, грубые черты лица, низкий грубый голос, шумное дыхание, частые ОРВИ. В 3-4 года возникают нарушения координации движений. Наблюдается прогрессирующая тугоухость, остеоартриты, узелковые поражения кожи спины.
При наличии синдрома Санфилиппо (III тип) ребенок в течение 3-5 лет нормально развивается, хотя иногда могут наблюдаться затрудненное глотание и неуклюжая походка. После трех лет у ребенка начинает развиваться апатия, возникает задержка психомоторного развития, нарушается речь, грубеют черты лица, возникает недержание кала и мочи, ребенок перестает узнавать окружающих. Такие дети умирают в возрасте 10-20 лет из-за интеркуррентных инфекций.
IV тип мукополисахаридоза характеризуется появлением первых симптомов в 1-3 года. Наблюдаются: резкая задержка роста, непропорциональное телосложение, грубые черты лица, кифоз или сколиоз, деформация грудной клетки; контрактуры в плечевых, локтевых, коленных суставах; плоскостопие; снижение мышечной силы; снижение слуха; дистрофия роговицы. Больные не доживают до 20-летнего возраста по причине сердечно-легочной недостаточности.
Для синдрома Шейе (V тип) характерен низкий рост, уплощенная переносица, короткая шея, контрактуры суставов, гипотония мышц конечностей, вегетативная лабильность, снижение сухожильных рефлексов, значительное помутнение роговиц.
Синдром Марото-Лами (VI тип) впервые проявляется после 2 лет. Он характеризуется отставанием в росте, грубыми чертами лица, бочкообразной грудной клеткой, малыми размерами верхней челюсти, короткой шеей, сгибательными контрактурами суставов рук. Интеллект больных не страдает.
Мукополисахаридоз VII типа устанавливают лишь при детальном биохимическом исследовании.
Синдром Ди Ферранте (VIII тип) схож с синдромом Моркио, но отличается выраженной задержкой интеллектуального и психомоторного развития.
Диагностика мукополисахаридоза
Диагностика мукополисахаридоза основывается на его характерных проявлениях, результатах рентгенологического исследования, установления экскреции гликозаминогликанов с мочой, исследования активности ферментов в фибробластах кожи.
Диагностировать мукополисахаридоз можно еще до рождения ребенка, используя для анализа амниотическую жидкость или ворсины хориона.
Лечение мукополисахаридоза
В настоящее время специфического лечения мукополисахаридоза не разработано.
Как правило, терапия мукополисахаридоза носит симптоматический характер. Пациента при этом наблюдают врачи различных специализаций: педиатр (по причине частых острых респираторных вирусных инфекций), хирург (в связи с наличием грыж), ортопед (в связи с нарушениями опорно-двигательного аппарата), отоларинголог (в связи с хроническими синуситами и отитами, нарушениями слуха), офтальмолог, невропатолог, нейрохирург.
В лечении мукополисахаридоза используются различные гормональные препараты (глюкокортикоиды, кортикотропин, тиреоидин), витамин А, цитостатики, сердечные препараты, переливания лейкоцитарной массы, крови, плазмы. Но все это вызывает лишь временные улучшения.
При синдроме Гурлера может проводиться трансплантация костного мозга. При ранних сроках ее проведения (до 1,5 лет) она может в значительной мере улучшить прогноз болезни, но данная процедура может вызывать ряд осложнений.
В США, странах Европы и Японии для лечения мягких форм мукополисахаридоза I типа применяется препарат альдуразим, который замещает недостающий в организме пациента фермент и приводит к улучшению состояния дыхательной системы, костей и суставов. Но внутривенное введение препарата не останавливает поражения ЦНС, поскольку альдуразим не проникает из крови в мозг. Данное средство применяется в случаях, когда трансплантация невозможна или непосредственно перед трансплантацией костного мозга, чтобы улучшить состояния больного и замедлить развитие болезни.
Таким образом, мукополисахаридоз – это редкое заболевание с неблагоприятным прогнозом для жизни пациента, поскольку с течением времени проявления болезни только усиливаются, а эффективного способа ее лечения пока нет. Единственный способ предотвратить заболевание – это обнаружить его еще в неонатальном периоде и предпринять соответствующие меры.