
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
- Код по МКБ-10
- Причины
- Симптомы
- Диагностика
- Что нужно обследовать?
- Как обследовать?
- Какие анализы необходимы?
- Лечение
- К кому обратиться?

Термин канальцевые дисфункции с гипокалиемией объединяет синдром Барттера (включая вариант Гительмана), псевдогипоальдостеронизм (синдром Лиддла) и пренатальный гиперпростагландин Е-синдром. Последний у взрослых не наблюдают.
 [1], [2], [3], [4], [5]
[1], [2], [3], [4], [5]
Код по МКБ-10
Причины синдрома Барттера
Синдром Барттера представляет собой генетически детерминированное заболевание, проявляющееся гипокалиемией, метаболическим алкалозом, гиперурикемией и повышением активности ренина и альдостерона.
Отдельно выделяют вариант Гительмана: помимо названных признаков, отмечают также гипомагниемию и гипокальциурию.
В настоящее время расшифрованы некоторые генетические механизмы синдрома Барттера и варианта Гительмана. Синдром Барттера наследуется по аутосомно-рецессивному, синдром Лиддла — по аутосомно-доминантному типу. Идентифицированы мутации, ответственные за развитие синдрома Лиддла (16р12.2-13.11 и 12р13.1).
Варианты синдрома Барттера
Тип I (неонатальный)
Фуросемид-, буметанидчувствительный Na + -, K + -, 2С1-транспортный белок восходящего колена петли Генле
АТФ-зависимый белок калиевого канала
Тиазидчувствительный транспортёр Na + и С1
[6], [7], [8], [9], [10], [11], [12]
Симптомы синдрома Барттера
Для синдрома Барттера, проявляющегося в раннем детском возрасте (неонатальный вариант), характерно тяжёлое течение с полиурией, дегидратацией, гипертермией, гиперкальциурией и ранним развитием кальциевого нефролитиаза.
Синдром Барттера, манифестировавший позднее (классический вариант), протекает более доброкачественно. Большинство пациентов начинают предъявлять жалобы в возрасте до 25 лет. Типичные симптомы синдрома Барттера — признаки гипокалиемии: мышечная слабость, парестезии, мышечные крампи вплоть до типичных судорог.
При выраженной гипокалиемии возможно развитие рабдомиолиза, осложняющегося острой почечной недостаточностью, однако подобные наблюдения редки. При синдроме Барттера артериальное давление остаётся нормальным, нередко наблюдают полиурию.
Вариант Гительмана часто впервые обнаруживают у взрослого человека. Гипомагниемия вызывает кальцификацию суставных хрящей, приводящую к постоянным артралгиям. Также наблюдают отложение кальция в склере и радужной оболочке глаза. Иногда развивается терминальная почечная недостаточность. У пациентов с вариантом Гительмана следует отдавать предпочтение постоянному амбулаторному перитонеальному диализу, сопряжённому с меньшим риском дальнейшего нарушения метаболизма электролитов.
Для синдрома Лиддла характерна выраженная артериальная гипертензия.
[13], [14], [15], [16], [17]
Диагностика синдрома Барттера
Лабораторная диагностика синдрома Барттера
Как при «классическом» синдроме Барттера, так и при варианте Гительмана отмечают значительное увеличение концентрации калия и хлоридов в моче.
При синдроме Лиддла наблюдают выраженное увеличение экскреции калия с одновременной задержкой натрия. Концентрация альдостерона в крови не изменена или снижена.
[18], [19]
Инструментальная диагностика синдрома Барттера
В отличие от синдрома Барттера, при варианте Гительмана в биоптатах почечной ткани не обнаруживают гиперплазии юкстагломерулярного аппарата.
Синдром Барттера

Синдром Барттера – это генетически обусловленная тубулопатия, проявляющаяся выраженными нарушениями электролитного обмена (гипокалиемией), кислотно-щелочного равновесия (метаболическим алкалозом), гиповолемией, компенсаторной гиперплазией юкстагломерулярного (околоклубочкового) аппарата почек и вторичным гиперальдостеронизмом. Диагностируется по клинической симптоматике: полиурии, отставании в психомоторном развитии, гипотонии мышц, а также лабораторным показателям крови и мочи. Лечение заключается в заместительной терапии препаратами калия, натрия и магния, приеме калийсберегающих диуретиков, ингибиторов синтеза простагландинов и АПФ.

- Причины
- Симптомы
- Диагностика
- Лечение синдрома Барттера
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Синдром Барттера в клинической урологии представляет собой редкую генную мутацию — дефект петли Генле, наследуемую по аутосомно-рецессивному типу и проявляющуюся, как правило, уже в детском возрасте. Неспособность почечных нефронов задерживать калий приводит к хронической потере его с мочой и уменьшению объема циркулирующей крови при нормальном или пониженном АД. В зависимости от вида пораженных генов различают: неонатальный синдром Барттера 1 и 2 типов, классический синдром Барттера, синдром Гительмана. Также встречается приобретенный синдром псевдо-Барттера, характеризующийся сходными проявлениями, но не сопровождающийся патологией почечных канальцев.
Причины
Причиной синдрома Барттера считают нарушение транспортной функции почечных канальцев, проявляющееся снижением реабсорбции ионов Cl (и, соответственно, Na) клетками восходящего отдела петли Генле. Это приводит к гиповолемии, избытку натрия и воды в дистальной части нефрона, усилению секреции ионов K и натрий-калиевого обмена. Гипокалиемия стимулирует, в свою очередь, образование простагландинов Е2 и I2, приводящее к усилению секреции ренина и ангиотензина II.
Хроническая гиперренинемия способствует развитию гиперплазии юкстагломерулярного аппарата почек и повышенной продукции альдостерона надпочечниками. Ангиотензин II и альдостерон вызывают увеличение уровня почечного калликреина с дальнейшим повышением содержания брадикинина плазмы крови. Альдостерон приводит к усилению выведения калия почками. Калликреин (брадикинин) и простагландины блокируют вазопрессорный эффект ангиотензина II, поддерживая нормальную величину артериального давления.
Синдром псевдо-Барттера может быть вызван продолжительным приемом диуретиков, длительной хлордефицитной диетой, периодически возникающей рвотой, чрезмерным приемом слабительных, муковисцидозом.
Симптомы
Синдром Барттера проявляется сразу после рождения или в раннем детском возрасте. Его клиническая картина обусловлена имеющимся хроническим дефицитом калия. Наблюдается полиурия и, как следствие, эксикоз (обезвоживание), поражение мышечной системы (слабость скелетных мышц, сердечной мышцы, гладкой мускулатуры, вялый псевдопаралич, судороги), отставание ребенка в умственном и физическом развитии, поражение нервной системы (парестезии и ригидность конечностей) при отсутствии артериальной гипертензии (нормальном или сниженном АД).
Неонатальный вариант патологии манифестирует в период внутриутробного развития плода многоводием, часто сопровождается преждевременными родами и имеет тяжелое течение. У недоношенных новорожденных наблюдается плохой аппетит, сонливость, быстрая потеря веса, задержка психомоторного развития, мышечная гипотония, нарушения зрения и слуха, гипертермия.
Классический тип синдрома проявляется в раннем детском возрасте (после 1 года жизни) задержкой роста и развития ребенка, полиурией, склонностью к дегидратации, рвотой, запорами, полидипсией. Синдром Гительмана выявляется примерно с 6-летнего возраста или позднее; характеризуется мышечной слабостью, утомляемостью, случаями возвратной тетании и имеет более доброкачественное течение.
При синдроме псевдо-Барттера развиваются аналогичные симптомы, обусловленные гипокалиемическим метаболическим алкалозом; данная патология часто встречается у молодых девушек, использующих для похудания диуретики и строго ограниченную диету.
Диагностика
Диагноз синдрома Барттера обычно устанавливается детским урологом по клинической симптоматике — сочетанию полиурии с мышечной гипотонией. К лабораторно-диагностическим критериям можно отнести низкую концентрацию ионов K, Cl, Na, Mg в сыворотке крови и их повышенное содержание в моче, гиперкальциурию, гиперфосфатемию, а также значительный уровень ренина и альдостерона плазмы крови, усиленную экскрецию простагландинов и калликреина с мочой, отсутствие артериальной гипертензии.
Неонатальный тип синдрома на первой неделе жизни можно определить по наличию метаболического алкалоза с гипокалиемией, низкому удельному весу мочи, содержащей большое количество ионов K, Na, Cl, Ca, высокому уровню простагландинов в крови и моче, большой активности ренина и альдостерона в крови.
При классическом варианте течения выявляют гипокалиемический метаболический алкалоз с повышенным или нормальным содержанием кальция, не нарушенную способность концентрировать мочу. В случае синдрома Гительмана обнаруживается резко выраженная гипомагниемия и гипокальциурия. По этим показателям синдром Барттера диагностируется при исключении приема диуретиков и слабительных средств, потерь калия и хлоридов через ЖКТ.
В редких случаях возможно выполнение биопсии почки, которая позволяет выявить гиперплазию околоклубочкового аппарата. Патологию следует дифференцировать от хронической рвоты, злоупотребления мочегонными препаратами, состояний, связанных с дефицитом магния, изолированного гиперальдостеронизма, хронической надпочечниковой недостаточности.
Лечение синдрома Барттера
Традиционное лечение различных типов синдрома включает заместительную и медикаментозную терапию. Необходимо обеспечение достаточного поступления калия и хлорида натрия с пищей, дополнительный прием препаратов калия. В лечении неонатального вида патологии сразу же после рождения ребенка начинают экстренную интенсивную заместительную терапию с помощью инфузий солевых растворов (NaCl, KCl). Для уменьшения потери калия организмом назначают калийсберегающие диуретики (спиронолактон, триамтерен, амилорид).
Необходим прием ингибиторов синтеза простагландинов (НПВС: индометацина, аспирина) и ингибиторов АПФ (каптоприла), снижающих секрецию ренина и альдостерона. У недоношенных младенцев из-за побочного действия индометацина его применение необходимо отстрочить до достижения детьми 4-6 недельного возраста. Коррекцию гипомагниемии при синдроме Гительмана проводят препаратами магния. Для лечения синдрома псевдо-Барттера необходимо устранить первопричину заболевания.
Прогноз и профилактика
Ранняя диагностика и адекватное лечение классического синдрома Барттера позволяет уменьшить тяжесть проявлений, отставание в умственном и физическом развитии. При неонатальном типе заболевания в отсутствии своевременного лечения возможна гибель ребенка из-за тяжелых электролитных нарушений и дегидратации организма. При тяжелом и долгом клиническом течении заболевания часто развивается нефрокальциноз, который может привести к хронической почечной недостаточности. Профилактика не разработана.
Синдром Барттера
Синдром Барттера — заболевание, которое передается на генетическом уровне и проявляется в виде гипокалиемии (нарушение электролитных обменных процессов), гиповолемии, метаболического алкалоза (сбой кислотно-щелочного равновесия), вторичного гиперальдостеронизма и компенсаторной гиперплазии юкстагломерулярного отдела почек.
- Причины синдрома Барттера
- Симптомы синдрома Барттера
- Диагностика синдрома Барттера
- Лечение синдрома Барттера
- Прогноз синдрома Барттера
Синдором Барттера проявляется с самого рождения, а при ухудшении состояния наблюдается развитие нефрокальциноза, что в итоге приводит к почечной недостаточности. Обнаружить данное заболевание можно по следующим признакам:
- нарушение психомоторного развития у ребенка;
- гипотония мышц;
- полиурия;
- в ходе анализа результатов мочи и крови.
В ходе урологических исследований специалисты заметили, что синдром Барттера является генной мутацией дефекта петли Генле. Это в своем роде наследственность согласно аутосомно-рецессивному типу. Почечные нефроны при синдроме Баттера не способны удерживать калий, поэтому он быстро выводится из организма вместе с мочой, а циркуляция крови при этом значительно уменьшается в объеме как при пониженном, так и при нормальном АД.
Синдром Барттера классифицируют по видам согласно пораженным генам, в зависимости от характеристик их делят на:
- 1-й тип неонатального синдрома Барттера;
- 2-й тип неонатального синдрома Барттера;
- 3-й тип — классическая форма заболевания;
- 4-й тип с нейросенсорной тугоухостью;
- 5-й тип — подобный аутосомно-доминантной гипокальциемии;
- 6-й тип — синдром Гительмана (сопровождается гипомагниемией и гипокальциурием).
Причины синдрома Барттера
Главная причина возникновения синдрома Барттера заключается в неправильности функционирования почечных канальцев, а именно процессов транспортизации. Такое нарушение проявляется через сокращение клетками восходящей части петли Генле реасорбции ионов Cl и Na, это поводит к:
- усилению калий-натриевого обмена;
- переизбытку в дистанционном отделе нефрона воды и натрия;
- интенсификации секреции К-ионов;
- гиповолемии.
Встречается также синдором псевдо-Барттера, но он никак не связан с генетическими мутациями, а вызван муковисцидозом, продолжительным диетическим питанием, длительным приемом слабительных препаратов и диуретиков.
Следственные связи причин синдрома Барттера и их особенности:
- Усиление секреции ангиотензина-2 и ренина становится последствием стимуляции гипокалиемии, способствующей формированию простагландинов I2, а также Е2.
- Повышенная выработка надпочечниками продуктов альдостерона и развитие гиперплазии юкстагломерулярным отделом почек — итог хронической гиперренинемии.
- Альдостерон и ангиотензин-2 являются стимуляторами роста составной части почечного калликреина, что становится причиной поднятия брадикинина, содержащегося в плазме крови.
- Калий быстро выводится почками из-за наличия альдостерона.
- Блокировка вазопрессорного действия ангиотензина-2 осуществляется простагландинами и калликреином, это служит поддержкой для обеспечения нормальных показателей артериального давления.
Симптомы синдрома Барттера
Симптоматика синдрома Баттера обнаруживается специалистами сразу после рождения или в первые года жизни ребенка. Заболевание характеризуется хронической нехваткой калия в организме, так как он быстро выводится. Основные симптомы следующие:
- Полиурия, приводящая к обезвоживанию (эксикозу);
- Общее поражение всей мышечной системы (частые судороги, слабый псевдопаралич, вялость сердечной мышцы, мышц скелета и гладкой мускулатуры);
- Физическое и умственное недоразвитие;
- Фригидность конечностей и парестезия, они выступают непосредственными показателями нарушений нервной системы.
Касательно типов синдрома Барттера, то неонатальный возникает еще в период внутриутробного развития при многоводии. Беременность женщины, у которой плод имеет данное заболевание, протекает достаточно сложно и чаще всего оканчивается преждевременными родами. В большинстве случаев у детей врачи отмечают сонливость, отсутствие аппетита, плохой набор веса или его потерю, мышечную гипотонию, гипертермию, нарушение психомоторного развития, слуха и зрения.
Классический тип синдрома Барттера начинает проявляться после 1-го года жизни в виде:
- задержки роста и развития;
- полиурии;
- полидипсии;
- нарушений пищеварительной системы (рвота, запор);
- склонности к дегидратации.
Последний 6-й тип (синдром Гительмана) синдрома Барттера становится явным после шестилетнего возраста, при этом у ребенка наблюдается сильная утомляемость, мышечная слабость, однако, течение заболевания отличается большей мягкостью, нежели у других типов.
Диагностика синдрома Барттера
Диагностируют синдром Барттера согласно присутствию мышечной гипотонии и полиурии в клинической картине пациента еще в детском возрасте. Проявление заболевания заключается в следующем:
- низкой концентрации в сыворотке крови ионов Na, Mg, K, Cl;
- повышенном содержании в моче ионов Na, Mg, K, Cl;
- гиперфосфатемии;
- гиперкальциурии;
- отсутствии гипертензии (артериальной);
- увеличении в плазме крови уровня альдостерона и ренина;
- экскреции калликреина и простагландинов.
Лабораторные анализы и проведение врачебной диагностики организма дают возможность убедиться в наличии или отсутствии у пациента синдрома Барттера, в редких случаях проводят биопсию почек. Она позволит проявиться гиперплазии в области околоклубочкового аппарата. Не стоит путать синдром Барттера с последствиями, связанными с частым употреблением препаратов мочегонного действия, хронической рвотой, дефицитом магния, надпочечниковой недостаточностью и гиперальдостеронизмом изолированного типа.
Лечение синдрома Барттера
При лечении синдрома Барттера применяют как медикаментозную, так и заместительную терапию. Главной задачей врачей будет обеспечение пациента необходимым количеством хлорида натрия и калия. Важно соблюдать диету, обогащенную вышеуказанными веществами и специальными препаратами.
К заместительной интенсивной терапии неонатального типа синдрома Барттера приступают мгновенно после рождения, при этом врачи используют инфузии солевых растворов и калиесберегающие диуретики. Важно включить ингибиторы синтеза простагландинов и АПФ, ведь это позволит понизить секрецию альдостерона и ренина. При недоношенности новорожденного применение индометацина нужно отсрочить на 4-6 недель, так как появления сложных побочных эффектов не избежать. Для коррекционного лечения синдрома Гительмана используют препараты магния, а при синдроме псевдо-Барттера специалисты выявляют основные причины и устраняют их.
Прогноз синдрома Барттера
Классический синдром Барттера невозможно вылечить полностью, но при условии проведения ранней диагностики и прохождения эффективного курса терапии, проявления патологии значительно уменьшатся, например, физическое и умственное развитие ребенка не будет столь отсталым. При неонатальном типе заболевания нельзя терять ни минуты, так как отсутствие своевременного лечения приведет в необратимым последствиям и гибели новорожденного. Смерть в таких случаях наступает по причине дегидратации и электролитных нарушений сложных форм.
Если клиническое течение отличается длительностью и тяжелым состояние пациента, тогда синдром Барттера начинает сопровождаться нефрокальцинозом, который в большинстве случаев приводит к хронической недостаточности почек.
Синдром Барттера
Что такое синдром Барттера?
Синдром Барттера (нормотензивный гиперальдостеронизм) — это редкое наследственное заболевание, которое препятствует способности почек поглощать соли, калий, кальций и другие электролиты, что приводит к чрезмерной потере всех этих соединений в моче. Он характеризуется обезвоживанием, усталостью, спазмами, слабостью, ломкостью костей и уплотнением почек (гломерулосклероз).
Существует пять основных типов синдрома Барттера, четыре из которых могут начинаться до рождения (антенатально) и классическая форма, которая начинается в раннем детстве и имеет тенденцию быть менее серьезной.
Заболевание может быть диагностировано с помощью анализов крови и мочи и подтверждено генетическим тестированием. Лечение направлено на устранение симптомов в каждом конкретном случае. Лекарство от синдрома Барттера нет.
Болезнь названа в честь доктора Фредерика Барттера, который впервые описал синдром в 1962 году.
Симптомы и признаки

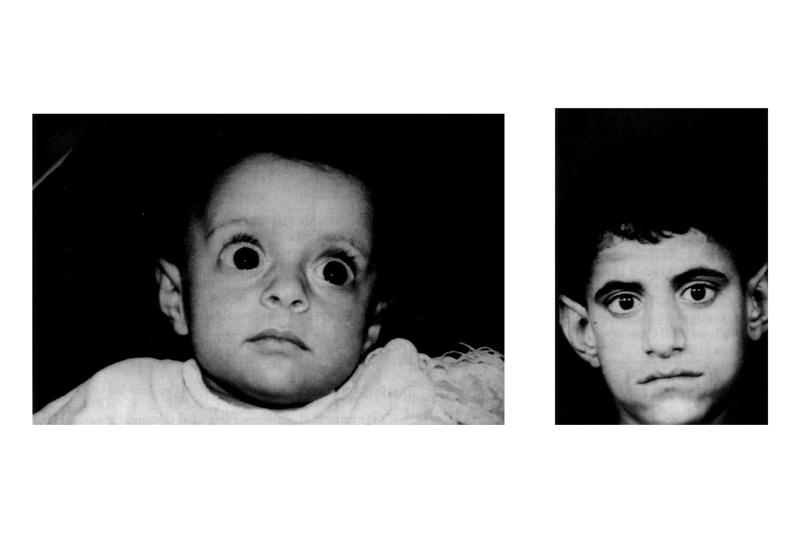
Симптомы синдрома Барттера связаны с чрезмерной потерей соли (хлорида натрия), калия и кальция в моче, каждая из которых имеет потенциально серьезные последствия. Вместе они характеризуют расстройство, при котором дети часто не могут расти и набирать вес с ожидаемой скоростью (так называемая неспособность развиваться, см. фото).
Среди характерных симптомов и признаков синдрома Барттера наблюдаются:
- Чрезмерная потеря соли, что может привести к обезвоживанию организма, запорам, жажде соли, усилению мочеиспускания (полиурия), усилению жажды (полидипсия) и пробуждению ночью к мочеиспусканию (никтурия).
- Чрезмерная потеря калия, что может привести к гипокалиемии (низкое содержание калия в крови), характеризующейся мышечной слабостью, судорогами, усталостью, учащенным сердцебиением (тахикардия), проблемами с дыханием, проблемами с пищеварением и потерей слуха.
- Чрезмерная потеря кальция в моче (гипокальциурия), что может препятствовать развитию костей у детей и вызывать ослабление костей (остеопения).
Симптомы могут существенно различаться, у некоторых детей будут только легкие симптомы. Те, у кого есть антенатальная форма, как правило, симптомы протекают хуже, главным образом потому, что потеря соли, калия или кальция может помешать нормальному развитию плода.
В тяжелых случаях признаки и симптомы синдрома Барттера будут видны до рождения. Нарушение функции почек может привести к накоплению околоплодных вод вокруг развивающегося плода (многоводия), вызывая преждевременные роды. Чрезмерное мочеиспускание у новорожденных часто может быть опасным для жизни. Также может возникнуть рвота и диарея.
Несмотря на тяжесть этих эпизодов, почечная функция детей может иногда нормализоваться в течение нескольких недель и не требует дальнейшего лечения.
Причины синдрома Барттера
Синдром Барттера представляет собой аутосомно-рецессивный паттерн, означающий, что для развития заболевания у ребенка должны присутствовать две копии аномального гена — одна от отца и одна от матери.
Синдром Барттера вызван мутациями одного из семи различных генов, каждый из которых относится к определенному типу синдрома Барттера. Дополнительные мутации могут привести к появлению подтипов с другим набором симптомов или серьезностью заболевания.
Гены предназначены для кодирования белков, которые транспортируют соль и электролиты, такие как калий и кальций, в почки для реабсорбции в петле Генле (U-образный канал, где вода и соль выделяются из мочи). Если гены мутируют, то полученные белки не могут транспортировать некоторые или все эти соединения через клетки петли Генле.
Специфические генетические мутации относятся к пяти основным типам синдрома Барттера:
| Имена | Тип | Генные мутации | Подробности |
| Антенатальный синдром Барттера | 1 | SLC12A1, NKCC2 | Имеет тенденцию быть серьезным с риском многоводия и преждевременных родов. |
| Антенатальный синдром Барттера | 2 | ROMK, KCNJ1 | Имеет тенденцию быть серьезным с риском возникновения многоводия и преждевременных родов. |
| Классический синдром Барттера | 3 | CLCNKB | Имеет тенденцию быть мягче, чем другие формы заболевания. |
| Синдром Барттерса с нейросенсорной глухотой | 4 | BNDS | Имеет тенденцию быть более серьезной, чем остальные формы, вызывает потерю слуха, в результате антенатального повреждения слухового нерва. |
| Синдром Барттера с аутосомно-доминантной гипокальциемией | 5 | CASR | Как правило, тяжелая форма болезни, затрагивающая в основном мальчиков и вызывающая задержки роста и остеопению. |
Синдром Барттера встречается редко, затрагивая лишь одного из 1,2 миллиона новорожденных. Чаще всего это происходит у детей, рожденных от родителей, состоящих в родстве (близких родственниках). Как представляется, это заболевание сильно распространено в Коста-Рике и Кувейте, чем среди любого другого населения.
Существует мало исследований, касающихся ожидаемой продолжительности жизни у детей с синдромом Барттера, но большинство данных свидетельствуют о том, что перспективы хорошие, если заболевание диагностировано и вылечено на ранней стадии.
Несмотря на влияние, которое синдром Барттера может оказать на почки, почечная недостаточность встречается редко.
Диагностика
Синдром Барттера диагностируется на основе исследования симптомов и истории болезни, а также различных анализов крови и мочи для выявления дисбаланса в уровнях соли, калия, кальция и других электролитов. Поскольку расстройство настолько редкое, почти всегда будет необходим вклад генетика, генетического консультанта и других специалистов.
Анализы крови, используемые для диагностики синдрома Барттера, позволяют выявить низкий уровень калия, хлорида, магния и бикарбоната в крови, а также повышенный уровень гормонов ренина и альдостерона.
Анализ мочи будет использоваться для выявления аномально высокого уровня натрия, хлорида, калия, кальция и магния в моче, а также наличия простагландина Е2 (маркера воспаления почек).
Антенатальные формы синдрома Барттера часто можно диагностировать до рождения, когда полигидрамниоз обнаруживается без наличия врожденных врожденных дефектов. Тут также будет повышенный уровень хлорида и альдостерона в амниотической жидкости.
Молекулярно-генетическое исследование может подтвердить диагноз. Есть несколько генетических тестов, которые могут обнаружить различные мутации, связанные с синдромом Барттера, доступные только через специализированные генетические лаборатории.
Может потребоваться дополнительное генетическое тестирование, чтобы дифференцировать синдром Барттера от тесно связанного, но более легкого наследственного расстройства, известного как синдром Гительмана.
Лечение синдрома Барттера
Основой лечения синдрома Барттера является восстановление баланса жидкостей и электролитов в организме. Подход может варьироваться от человека к человеку в зависимости от тяжести симптомов. Некоторым детям может потребоваться минимальные терапевтические вмешательства или самопроизвольная нормализация без необходимости дальнейшего лечения. Другим может потребоваться помощь в течение всей жизни от группы специалистов, включая педиатра, терапевта или нефролога.
Лекарственные препараты

Добавки натрия, хлорида калия и магния часто используются для коррекции электролитного дисбаланса. Для лечения воспаления и снижения уровня простагландина, которые способствуют чрезмерному мочеиспусканию, могут быть назначены другие препараты, включая нестероидные противовоспалительные препараты (НПВП), такие как Адвил (ибупрофен), Целебрекс (целекоксиб) или Индоколлир (индометацин).
Блокаторы желудочной кислоты, такие как Квамател (фамотидин), Тагамет (циметидин) или Зантак (ранитидин), могут быть необходимы для снижения риска возникновения язв и кровотечений, вызванных длительным применением НПВП.
Другие лекарства, такие как антагонисты альдостерона, блокаторы рецепторов ангиотензина II и ингибиторы ангиотензинпревращающего фермента (АПФ). может потребоваться для снижения уровня ренина и риска повреждения почек.
В зависимости от того, какие электролиты несбалансированы, некоторым людям могут потребоваться мочегонные средства, не содержащие калия, такие как спиронолактон или амилорид, для увеличения выделения натрия в моче, но удерживающие калий.
Другие вмешательства
Младенцам с тяжелыми, угрожающими жизни симптомами может потребоваться внутривенная (в/в) замена соли и воды. Дети, которые страдаю неспособность развиваться, часто получают выгоду от терапии гормоном роста, чтобы предотвратить задержку роста и низкий рост. Также могут быть использованы кохлеарные имплантаты для лечения глухоты, связанной с синдромом Барттера 4 типа.
В дополнение к добавкам, для увеличения потребления калия и соли будет адаптироваться питание больного. В дополнение к достаточной гидратации детей, поощряется употребление соленной пищи.
С возрастом синдром Барттера становится легче контролировать и лечить.
Прогноз для жизни
Синдромы Барттера являются аутосомно-рецессивными расстройством, и не поддается лечению. Степень нетрудоспособности зависит от тяжести дисфункции рецептора, но прогноз во многих случаях хороший, так как пациенты могут вести достаточно нормальный образ жизни.
Синдром Барттера: факторы риска развития, клинические проявления, диагностика и тактика лечения
Врожденные нарушения транспорта в почечных канальцах формируют спектр редких состояний, при каждом из которых поражаются определенные сегменты нефрона. Успехи генетики и молекулярной биологии позволили расшифровать патогенез многих таких заболеваний и углубили наши представления о регуляции водно-электролитного обмена в норме.
Синдром Бартера редкая форма гипокалиемического метаболического алкалоза с гиперкальциурией наследуется аутосомно-рецессивно. Различают два клинических подтипа синдрома Бартера. Антенатальный синдром Бартера (называемый также синдромом гиперпродукции простагландина Е) обычно проявляется у новорожденных и протекает тяжелее, чем классический синдром Бартера; он включает многоводие в анамнезе, потерю соли и выраженное обезвоживание.
Более легкий классический фенотип проявляется позднее задержкой развития ребенка и частыми эпизодами обезвоживания в анамнезе. Фенотипически сходный синдром Гительмана обусловлен другим генетическим дефектом. Описан также вариант антенатального синдрома Бартера с нейросенсорной глухотой и ХПН, имеющий другую генетическую основу.
Патогенез синдрома Бартера у детей. Биохимические сдвиги при синдроме Бартера (гипокалиемический метаболический алкалоз с гиперкальциурией) напоминают последствия применения петлевых диуретиков и отражают нарушение транспорта натрия, хлорида и калия в восходящем отделе петли Генле. Потеря натрия и хлорида, приводящая к уменьшению внутрисосудистого объема, стимулирует ренин-ангиотензин-альдостероновую систему.
Альдостерон усиливает реабсорбцию натрия и секрецию калия, тем самым усугубляя гипокалиемию. Он усиливает также секрецию ионов водорода в дистальных отделах нефрона, что усугубляет метаболический алкалоз. Гипокалиемия стимулирует синтез простагландинов, которые еще больше активируют ренин-ангиотензин-альдостероновую систему. В основе синдрома Бартера лежат три разных генетических дефекта транспортеров, функционирующих на уровне петли Генле.
Каждый из них тем или иным образом участвует в транспорте натрия и хлорида. При антенатальном синдроме Бартера обнаруживаются мутации гена, кодирующего натрий/калий/2 хлоридный транспортер NKCC2 (объект действия фуросемида), или гена люминальных калиевых каналов (ROMK), тогда как для классического синдрома характерны дефекты базолатеральных хлоридных каналов (ClC-Kb).
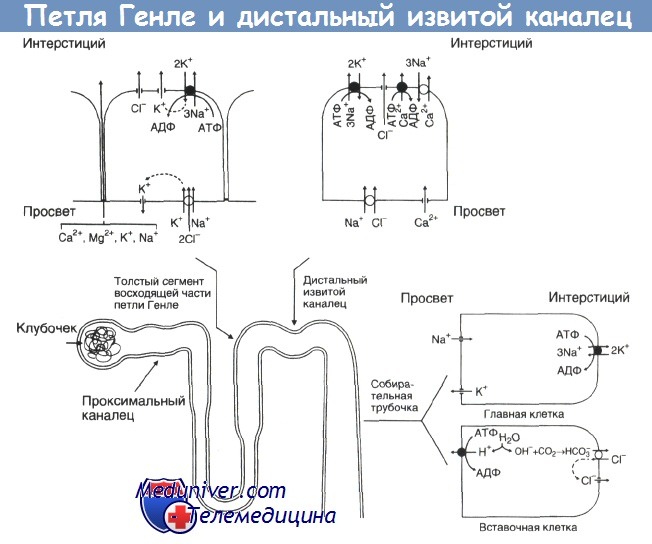 Основные переносчики апикальной и базолатеральной мембраны толстого сегмента восходящей части петли Генле, дистального извитого канальца и коркового отдела собирательной трубочки.
Основные переносчики апикальной и базолатеральной мембраны толстого сегмента восходящей части петли Генле, дистального извитого канальца и коркового отдела собирательной трубочки.
Клинические проявления синдрома Бартера у детей. Как выясняется из анамнеза, при беременности было многоводие. У новорожденного возможна дизморфия, например треугольное лицо, оттопыренные уши, косоглазие и опущение углов рта. Родственная связь между родителями указывает на аутосомно-рецессивное наследование синдрома. Для более позднего возраста характерны повторные эпизоды обезвоживания, задержка развития и биохимические нарушения. При синдроме Бартера всегда имеют место гипокалиемия и метаболический алкалоз.
Содержание кальция в моче обычно повышено. Часто значительно повышен уровень ренина, альдостерона и простагландина Е в сыворотке крови, особенно при тяжелой антенатальной форме синдрома. АД в большинстве случаев нормальное, хотя обезвоживание при выраженной потере соли у больных с антенатальной формой синдрома может приводить к артериальной гипотонии. Функция почек, как правило, сохранена. При УЗИ иногда обнаруживается нефрокальциноз как следствие гиперкальциурии.
Диагностика синдрома Бартера у детей. Диагноз устанавливают на основании клинической картины и лабораторных данных. У новорожденных о синдроме Бартера свидетельствует гипокалиемия (обычно ниже 2,5 ммоль/л) на фоне метаболического алкалоза. В типичных случаях отмечается гиперкальциурия. Гипомагниемию обнаруживают лишь у немногих больных; она более характерна для синдрома Гительмана. Поскольку проявления напоминают последствия продолжительного использования петлевых диуретиков, всегда необходимо выяснить, применялись ли эти средства (даже у маленьких детей).
Аналогичная клиническая картина имеет место при хронической рвоте, но при этом содержание хлорида в моче снижено, тогда как при синдроме Бартера оно повышено. При гистологическом исследовании почек находят гиперплазию юкстагломерулярного аппарата. Однако диагностическая биопсия при синдроме Бартера выполняется редко.
Лечение и прогноз синдрома Бартера у детей. Терапия синдрома Бартнера направлена на предотвращение обезвоживания и поддержание питания и коррекцию гипокалиемии. Зачастую требуются очень большие дозы калия, но и в этих случаях его уровень в сыворотке не всегда удается нормализовать, особенно у новорожденных. Грудные и маленькие дети могут нуждаться и в добавках натрия. Эффективен также индометацин, ингибирующий синтез простагландинов.
При внимательном отношении к электролитному балансу, объемному статусу и росту ребенка долговременный прогноз обычно благоприятный. Однако хроническая гипокалиемия, нефрокальциноз, длительное введение индометацина могут иногда приводить к развитию интерстициального нефрита и ХПН.











